

The presence of adapter sequences in next-generation sequencing (NGS) data significantly reduces the quality of assembling and other downstream analysis results. Therefore, in SeqSphere+ Trimmomatic (citation) can be used to perform a trimming of Illumina adapter sequences in FASTQ read data.
By default, adapter trimming is automatically performed in a pipeline if adapter sequences were found by FastQC with state 'warning' or 'failed'. It can also be disabled completely or it can be forced to be performed always and independently of FastQC.
When performed in the pipeline, Trimmomatic searches for all adapter sequences provided with SeqSphere (see below). After Trimmomatic was performed, the phrase 'adapters trimmed' is added to the procedure details field 'Assembler pre-processing'.
Tools Menu Function
Trimmomatic can also be performed manually by using the menu function Tools | Genome Utilities | Illumina Adapter Trimming (Trimmomatic).
When the function is invoked, a dialog windows is opened. In the left part of the dialog the available adapter sequences are listed. The Illumina adapter sequences for Nextera and TruSeq library prep kits are distributed together with Trimmomatic. Multiple or even all adapter sequences can be selected at once.
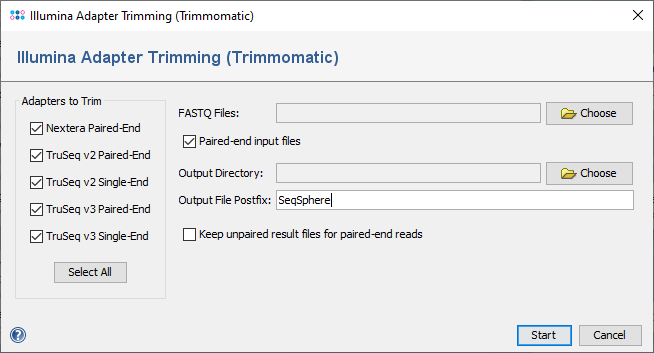
The following adapter sequences are provided:
- Nextera Paired-End
- TruSeq v2 Paired-End
- TruSeq v2 Single-End
- TruSeq v3 Paired-End
- TruSeq v3 Single-End
The dialog window has the following further fields/options in the right part:
- FASTQ Files
- One or multiple FASTQ file(s) (single-end or paired-end) can be selected here. Also, all selected input files should be single-end OR paired-end only and a mixture of both is not acceptable.
- Paired-end input files
- The input data will be considered as paired-end or single-end based on user selection of this option.
- Output Directory
- The directory where to store the trimmed FASTQ files must be selected here.
- Output File Postfix
- A postfix that is appended to the name of the trimmed FASTQ files can be defined here.
- Keep unpaired result files for paired-end reads
- For paired-end data reads that have no corresponding read in the FASTQ file of the other direction are removed from the trimmed FASTQ files. If this option is enabled, those reads are kept in an extra FASTQ file with the postfix "_Unpaired". This option is enabled only when Paired-end input files checkbox is selected.
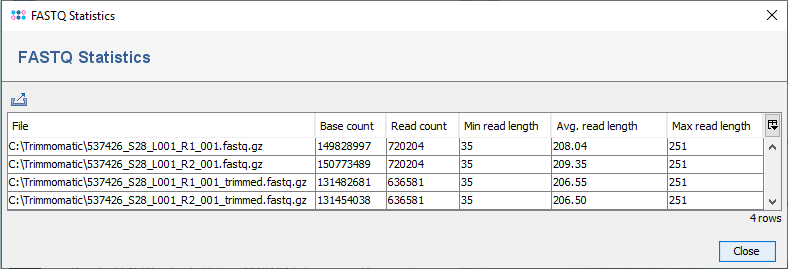
After Trimmomatic has finished a message dialog is shown. The button Show FASTQ Statistics in this dialog can be used to show the statistics (e.g., base count) for the input files and all created output files.
![]() Hint: Trimmomatic is run with the recommended default settings: seed mismatches=2, palindrome clip threshold = 30, and simple clip threshold = 10
Hint: Trimmomatic is run with the recommended default settings: seed mismatches=2, palindrome clip threshold = 30, and simple clip threshold = 10