

Contents
Introduction

The Staphylococcus aureus spa-typing task template can be used for WGS (from reads or pre-assembled) and for Sanger sequencing data to assign a type based on the repeat region of the Staphylococcus aureus protein A gene (spa). To guarantee a standardized nomenclature, the spa-types and repeats are named by a global nomenclature that is controlled by the Ridom SpaServer. They are named with an ID which is either a leading "t" (spa-type) or "r" (repeat) followed by a unique number.
The current version of the task template requires SeqSphere+ client version 8.3 (and server version 8.3) or later. It can be downloaded from the Task Template Sphere. Information on how to import a StaphType database can be found in the FAQ.
For Sanger sequencing data, the algorithm is exactly the same as in the former StaphType software. For WGS data, the spa-typing task tries to find the repeat region by searching for all known repeat sequences. If a repeat region is found, it is trimmed by the 5' and 3' signatures (RCAMCAAAA, TAYATGTCGT). Then the trimmed region is searched for known repeats and for potentially new repeats that are matching a specific pattern. If the known repeats are matching a spa-type, this spa-type is assigned. Finally the spa-typing result is QC controlled by determining a reliability rate (see table below). If multiple regions (e.g., on multiple contigs) with repeats are found in the first step, a spa-type is only called, if exactly one region has a sufficient spa-typing result, or if all regions have the same spa-type.
The Task Entry Overview for a spa-typing task shows a result message colored by the reliability, the four result fields, and potentially links to unreliable positions in the sequence that need to be checked.
Submitting New Spa-Types
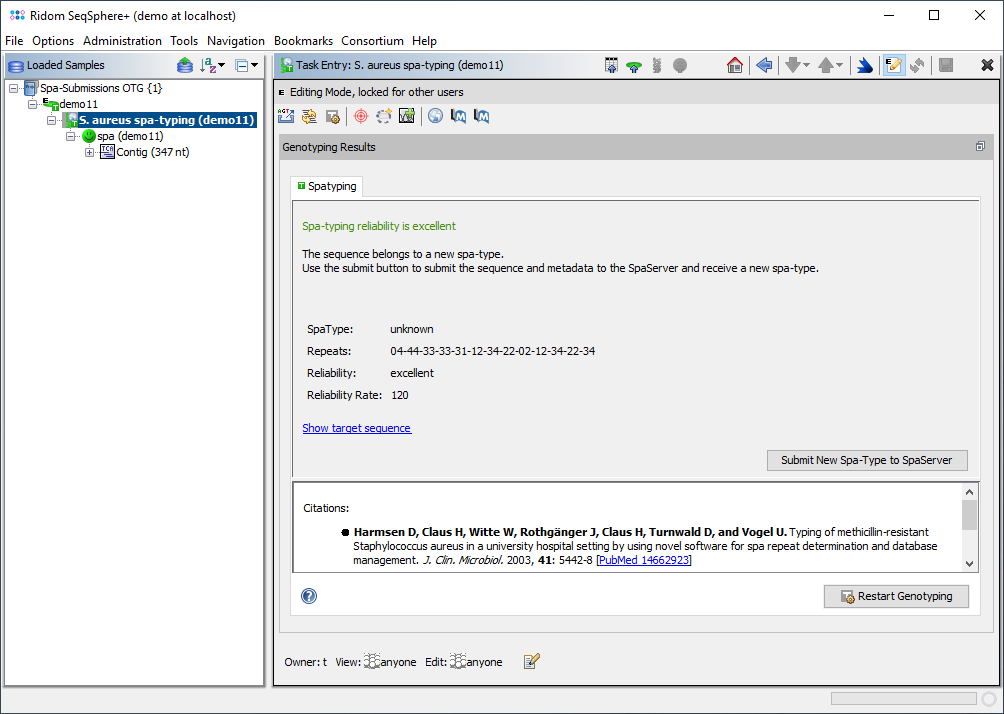
If an unknown spa-type is found and the reliability is "good" or "excellent" (reliability rate >= 95), the new spa-type can be submitted to the SpaServer by using the submission button in the Task Entry overview. Please note that with WGS data new spa-types that contain no new spa-repeat(s) can be submitted only. New spa-types that contain new spa-repeat(s) can be submitted with Sanger data only.
The following meta-data fields are submitted together with the sequence data and the spa-typing result:
- Submitter User ID (database ID and login name) *
- Submitter Email Address *
- Submitter Organisation *
- Submitter City *
- Submitter Country *
- Sample ID *
- Collection Date *
- Country of Isolation *
- Origin (Source Type and Source Subtype)
- MRSA/MSSA
* mandatory field
Result Fields
The S. aureus spa-typing provides four result fields:
- SpaType (shown in result table and used in comparison tables for distance calculation)
- Repeats (shown in result table)
- Reliabilty (shown in result table)
- Reliabilty Rate
Reliability Rating
The reliability of a spa-typing result can be poor, sufficient, good, or excellent. Submitting a strain for a new spa-type requires the reliability good or excellent (the latter can only be reached with sequence data Sanger chromatogram files).
Internally, the reliability is calculated as a numeric value between 0 and 120 with criteria shown in the table below.
| Reliability Rating | Input Data | Signature Search | Repeat Search | |
|---|---|---|---|---|
| 120 | excellent | as below | as below | as below, but less than 5 editing steps in repeats |
| 110 | good | Sanger sequencing data with consensus of two chromatograms | as below | as below, but no low-quality bases (consensus base quality < 20) |
| 100 | good | Sanger sequencing data with at least one chromatogram file | as below | as below, but no editing steps in new repeats |
| 95 | good | WGS data or Sanger sequencing data with at least one chromatogram file | as below | as below, but no new repeats |
| 90 | sufficient | Sanger sequencing data with at least one chromatogram file | as below | as below |
| 60 | sufficient | WGS data or consensus of two Sanger sequencing FASTA files | as below | as below |
| 50 | sufficient | as below | both signatures found on correct positions | as below |
| 40 | sufficient | as below | both signatures found, but position shifted less than 20nt from expected position | as below, but no high quality mismatches (chromatogram base quality≥15) |
| 30 | poor (not reliable) | as below | both signatures found, but position shifted | as below |
| 20 | poor (not reliable) | as below | at least one signature not found, but sequence to short | as below |
| 10 | poor (not reliable) | as below | at least one signature not found | as below, but not more than 5 low-quality positions (consensus base quality < 20) |
| 5 | poor (not reliable) | as below | n/a | continuous repeat succession without gaps |
| 0 | poor (not analyzable) | any FASTA sequence | n/a | no repeats or noncontinuous |
Early Warning Alert
Early Warning Alerts (EWA) are defined per project. They are used to automatically detect for newly processed samples (query samples) samples with the same spa-type (hit samples) that are already stored in the project. If samples within the defined thresholds are found, an EWA is triggered and stored in the database. - The spa Early Warning Alert definition can be created and managed by pressing the button Add Early Warning Alert Definition in the project editor below the task templates section. The button is only enabled if a spa or cgMLST Task Template was added to the project. The button icon is grey if no EW-Alerts are defined, else it is colored red. An EWA definition contains several sections. All sections are identical with the sections used for defining a cgMLST EWAs except the Allelic Profile Distance Criteria section that is replaced against Spa-Type Distance Criteria section.
Search Similar Samples
The search for similar samples can be used to find for selected samples other samples in the database, that have the same or a similar spa-type. Two spa-types are regarded as similar if the BURP distance is equal or below 4. Furthermore, only spa-types with at least five repeats can be compared for similarity (for details see [PubMed 17967176]). - If a S. aureus spa-typing task template is chosen for distance calculation, the Allelic Profile Distance Criteria section in the general search for similar samples dialog is replaced against a Spa-Type Distance Criteria section. Here it can be chosen if samples with the same or with a similar spa-type should be searched.
Comparison Table
When the SpaType field is used in a Comparison Table, distances for this field are calculated using the BURP (Based Upon Repeat Patterns) alignment algorithm. BURP distances can not be combined with distances from other task templates. Furthermore, distances are only calculated for spa-types with at least five repeats and therefore shorter spa-types are excluded from analysis. A detailed description of the BURP algorithm can be found in the following two publications:
Mellmann A, Weniger T, Berssenbrügge C, Rothgänger J, Sammeth M, Stoye J, and Harmsen D. Based Upon Repeat Pattern (BURP): an algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol. 2007, 7: 98 [PubMed 17967176] and Sammeth M, and Stoye J. Comparing tandem repeats with duplications and excisions of variable degree. IEEE/ACM Trans Comput Biol Bioinform. 2006, 3: 395-407 [PubMed 17085848].
When drawing a minimum spanning tree (MST) the samples are clustered by shading into MST clusters. By default all samples that have a BURP distance of 4 or less to each other are highlighted by this means. Therefore, these clusters correspond to the previous StaphType 'BURP CCs' except that CC founders no longer name the cluster. The settings for the MST clusters can be modified in the first tab of the MST Options.