

Contents
Overview
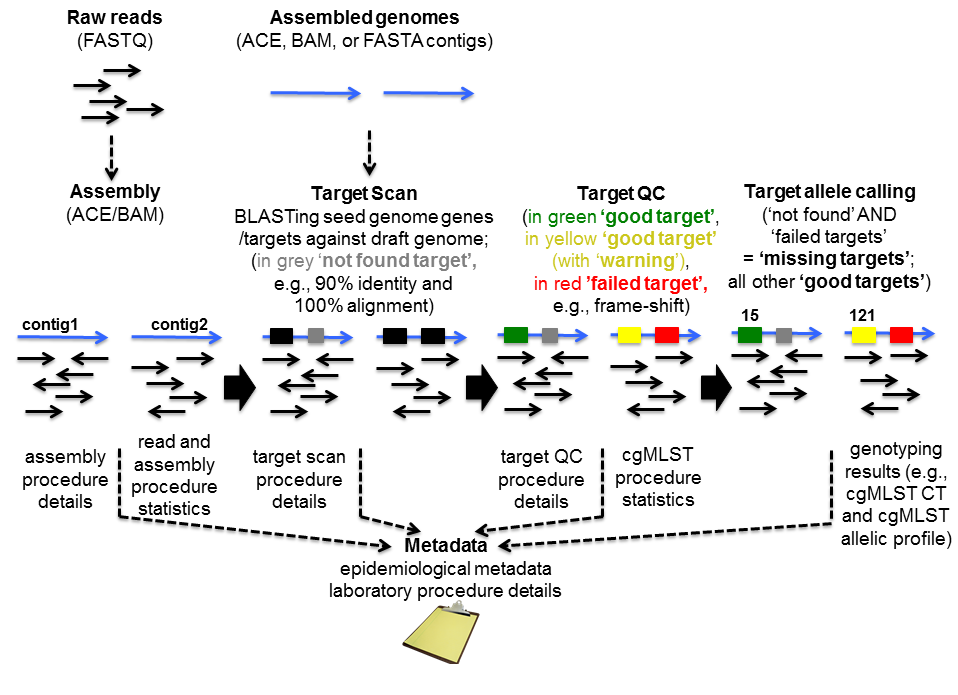
Assembly and consensus sequence calling
Assembling with Velvet
For de novo assembly done with the MS Windows SeqSphere+ Client the FASTQ files are first quality-trimmed (default setting for trimming is to trim on both ends of the reads until the average base quality is >30 in a window of 20 bases), downsampled (for Illumina downsampling to approximately 120x coverage of the expected genome size is default) and then de novo assembled using the Velvet assembler (version 1.1.04) with k-mer optimization procedure (optimum criteria is average length of contigs with more than 1000 bp). The AMOS file that is created by Velvet is automatically converted to ACE file format containing all mapped reads. The consensus bases are newly assigned by a SeqSphere+ consensus caller and stored in the ACE file. Most notable the consensus caller calls a ‘N’ for a position if a base is not supported by 60% or more of the reads at this position.
Assembling with SKESA
For de novo assembly done with the Linux version of SeqSphere+ Client (version 9.0) or with the Windows version on Windows 10 with installed WSL the SKESA assembler (version 2.4.0) can be used. Here quality-trimming of reads is in contrast to downsampling not done by SeqSphere+ by default. SKESA is run with recommended default parameters. By default, a polishing step is performed after assembling: reads are mapped back to the resulting FASTA contig file and an additional SeqSphere+ consensus calling is done.
Assembling with SPAdes
For de novo assembly done with the Linux version of SeqSphere+ Client (version 9.0) the SPAdes assembler (version 3.15.4) can be used. Here quality-trimming of reads is in contrast to downsampling not done by SeqSphere+ by default. SPAdes is run with recommended default parameters (slightly different for various sequencing platforms). By default, a polishing step is performed after assembling: reads are mapped back to the resulting FASTA contig file and an additional SeqSphere+ consensus calling is done.
Mapping with BWA
Currently reference-assisted assembly (‘mapping’) is only applied for Mycobacterium tuberculosis cgMLST. Here the BWA-SW algorithm (version 0.6.1/0.6.2 for Windows and Linux respectively) or BWA-MEM (version 0.7) is used. SeqSphere+ quality-trimming of reads is disabled by default. Reads are downsampled to approximately 120x coverage of the expected genome size before performing the BWA-SW mapping with default parameters. The above described SeqSphere+ consensus caller reads the resulting BAM file and calls a consensus sequence by applying the same majority rule as above. If the reference genome has regions that are uncovered in the genome under study, those gaps are deleted in the resulting consensus sequence and juxtapositional covered regions are concatenated. This procedure applies also for FASTA consensus files from mappings that are exported from SeqSphere+.
Target scan, quality control, and allele calling
Finding, evaluating, and allele calling the core and accessory genome genes (targets) defined in the schemes is for each target separately a three-step procedure, i.e. first ‘BLASTing’ and aligning, second applying some quality criteria for the found hit, and finally assigning a known or new allele type integer. The input genome file format may be ACE, BAM, GenBank, or FASTA.
Target scan
A BLASTN (BLAST version 2.2.12 or BLAST+ version 2.9.0+ if seed genome >= 6 Mbases) similarity search (word size: 11, mismatch penalty: -1, match reward: 1, gap open costs: 5, and gap extension costs: 2) is performed using the scheme’s target sequences of the seed genome (ref.-seqs.) as query sequences and the to be compared genome contig(s) as subject sequence(s). For each target a separate BLAST hit list is produced. To speed up the ‘blasting’ all contigs shorter than 200 bases are ignored as subject sequences by default. All BLAST hits with an alignment that ends near the start and/or end of a query sequence (less than 16 bases) are extended. Thereby the missing bases at the start and/or end are extracted from the query and the subject sequence, and are aligned separately using a global alignment with affine gap score (gap open costs: 4 and gap extension costs: 3). The resulting alignment(s) are then concatenated to the original alignment of the BLAST hit. Next the percentages of identity and alignment to the ref.-seq. are calculated for all extended and the other not-extended BLAST hits. All hits that pass the configurable thresholds (90% identity and 100% alignment by default for SeqSphere+ cgMLST schemes; Fig. 2) are marked as top-matching hits. The required identity threshold is somewhat species-specific (e.g., for the SeqSphere+ C. difficile cgMLST scheme only 80% identity is required). If only one top-matching hit is found this hit is selected for further processing. If multiple top-matching hits are found then no hit is selected to avoid ambiguous results (it is tried to eliminate those targets already during the scheme definition process). If no hit is selected for a target it is called in the software a ‘not found target’. The subject sequences of all selected top-matching hits are extended by 10 bases at each end. Those sequences are next aligned to each corresponding target ref.-seq. using a heuristic alignment that first applies BLASTN to find the longest overlapping region and then uses banded alignments to align the sequences left and right of the hit (bandwidth: 100, gap opening penalty: 6, and gap extension penalty: 3). Those determined overlap with the ref.-seq. defines the target sequence.
Optionally it can be configured that all known allele sequences for each target are searched, if no selected hit was found for a target in the ref.-seq. blasting step (or if no ref.-seq. is configured for a target in the scheme). In this case all known alleles for each target are used as query sequences instead of the target sequences of the seed genome only (option not applied for SeqSphere+ cgMLST schemes as it is quite compute-intensive and more importantly may produce non-reproducible results when a sample is repeatedly queried against an expanding allele nomenclature database; however default setting of BIGSdb). The multiple query sequences per target produce one BLAST hit list. Some hits are extended and thresholds are applied as described above. If only one top-matching hit is found this hit is selected for further processing. If multiple top-matching hits are found for a target, but only one has 100% identity with the corresponding query sequence, this hit is selected. Else, if multiple top-hits are found that are all overlapping each other in the subject sequence, the best hit (highest bit-score) is selected. Else, if multiple top-hits are found that are not all overlapping, no hit is selected. All further processing is done exactly as described above. In case it is known that there are two or more very similar genes present and queried in the subject genome it can be configured in the software, that always the best hit (highest bit-score) should be selected if multiple top-matching hits are found (option never applied for SeqSphere+ cgMLST schemes).
Target quality control

In the second step, the core and accessory genome target sequences are assessed for quality. In SeqSphere+ currently implemented quality control (QC) options are shown in Fig. 3. The various QC criteria can be ignored or configured to produce an ‘Error’ or just a ‘Warning’. If a target sequence produces an error it is not used in further downstream analysis (within the software this is called a ‘failed target’ and the target is colored in grey; ‘not found’ and ‘failed targets’ together are called ‘missing targets’). A target with a warning that is colored in yellow within the software is used further on; it is just flagged to indicate the user that there might be a problem with that target. A target producing neither an ‘Error’ nor a ‘Warning’ is colored in green within the software.
By default SeqSphere+ public cgMLST schemes produce errors for the following four criteria: (1) if the length of target sequence does not equal the reference sequence length plus or minus three triplets; (2) if there is any ambiguous base (e.g., N) in the target consensus sequence; (3) if there is a ‘frameshift’ detected, i.e. the number of insertion(s) of the target consensus sequence in comparison to the reference sequence minus the number of deletion(s) must be dividable by 3; and (4) if the start and stop codons are found on correct positions and no internal stop codons exist. SeqSphere+ supports all NCBI translation tables and for every scheme one specific translation table is defined.
SeqSphere+ tests for minimum coverage (this is not the average coverage along the target but the minimum coverage for every position in the target sequence). Currently only a warning is elicited if the coverage is low (default setting is less than 5 reads per target position; the idea is that ideally FASTQ, ACE, and FASTA contig [exported from SeqSphere+] files produce exactly the same allele calls). SeqSphere+ knows but currently does not evaluate the orientation of the WGS reads (e.g., every position in a target sequence must be covered by at least one forward and reverse read; this option is currently only available for Sanger sequencing data).
Target allele calling
Finally, only if a target passed successfully both previous steps it is considered a 'good target' and assigned either a new or existing numerical allele type. For finding existing alleles with associated numerical allele type a simple perfect matching string search of all known target allele sequences is done. If no perfect match is found a new allele type is assigned. The combination of all core and/or accessory genome ‘good’ alleles in each sample forms an allelic profile that is used to calculate distance matrices. Distance matrices are finally used to draw either minimum-spanning or neighbor-joining trees.
Therefore, like MLST, the genome-wide gene-by-gene allele calling cgMLST approach uses alleles as the unit of comparison, rather than nucleotide sequences. In allele-based comparisons among isolates, each allelic change is counted as a single genetic event, regardless of the number of nucleotide polymorphisms involved. This provides a simple and effective correction for the fact that in many bacteria, common horizontal genetic transfer events account for many more polymorphisms among specimens than rarer point mutations. The cgMLST approach retains information at all loci and avoids the need to categorize which changes are recent point mutations and which are due to recombination. As core and accessory genome MLST schemes record the sequences of allelic variants, those schemes can also be used for sequence-based analyses (e.g., SNP calling or using concatenated sequences) when this is appropriate (adapted from: Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. MLST revisited: the gene-by-gene approach to bacterial genomics. Nature Rev Microbiol. 2013, 11: 728-36 [PubMed 23979428]).
Metadata
During the whole procedure metadata are produced and stored (procedure details, procedure statistics, and genotyping results).